

Kapitel 3
Vilken metod ska man välja?
I föregående text har du kunnat läsa om en rad olika metoder som används för att analysera både etablerade och nya biomarkörer. Här kan du läsa mer om vilken analys som är lämplig att använda.
Det mänskliga genomet
Det exakta antalet gener i det mänskliga genomet är fortfarande ett ämne för diskussion men man brukar säga att det handlar om cirka 20 000 gener. Samlingsnamnet för denna del av arvsmassan är exomet, det vill säga alla kodande regioner av genomet. Exomet motsvarar endast cirka en procent av hela genomet. Resten, som kallas de icke-kodande delarna av genomet, utgörs till viss del av reglerande områden som i den enskilda cellen hjälper till att styra huruvida gener ska uttryckas eller ej. Men stora områden utgörs av DNA som man ännu inte vet vad det har för funktion. Det kallades tidigare för ”skräp-DNA” men numera vet man att detta DNA fyller många viktiga funktioner, bland annat genom att reglera geners aktivitet och påverka hur proteinkodande regioner tolkas.
Vad ska analyseras?
Eftersom olika molekylära metoder har olika styrkor och svagheter behöver man först bestämma sig för vad man behöver identifiera. Gäller det en enskild mutation eller många olika typer av avvikelser i många gener? Förväntar man sig små mutationer på basparsnivå eller stora komplexa avvikelser som translokationer? Hur känslig behöver analysen vara för att man ska känna sig trygg med ett negativt svar? Här beskrivs hur man kan resonera kring val av några av de vanligare molekylära metoderna.

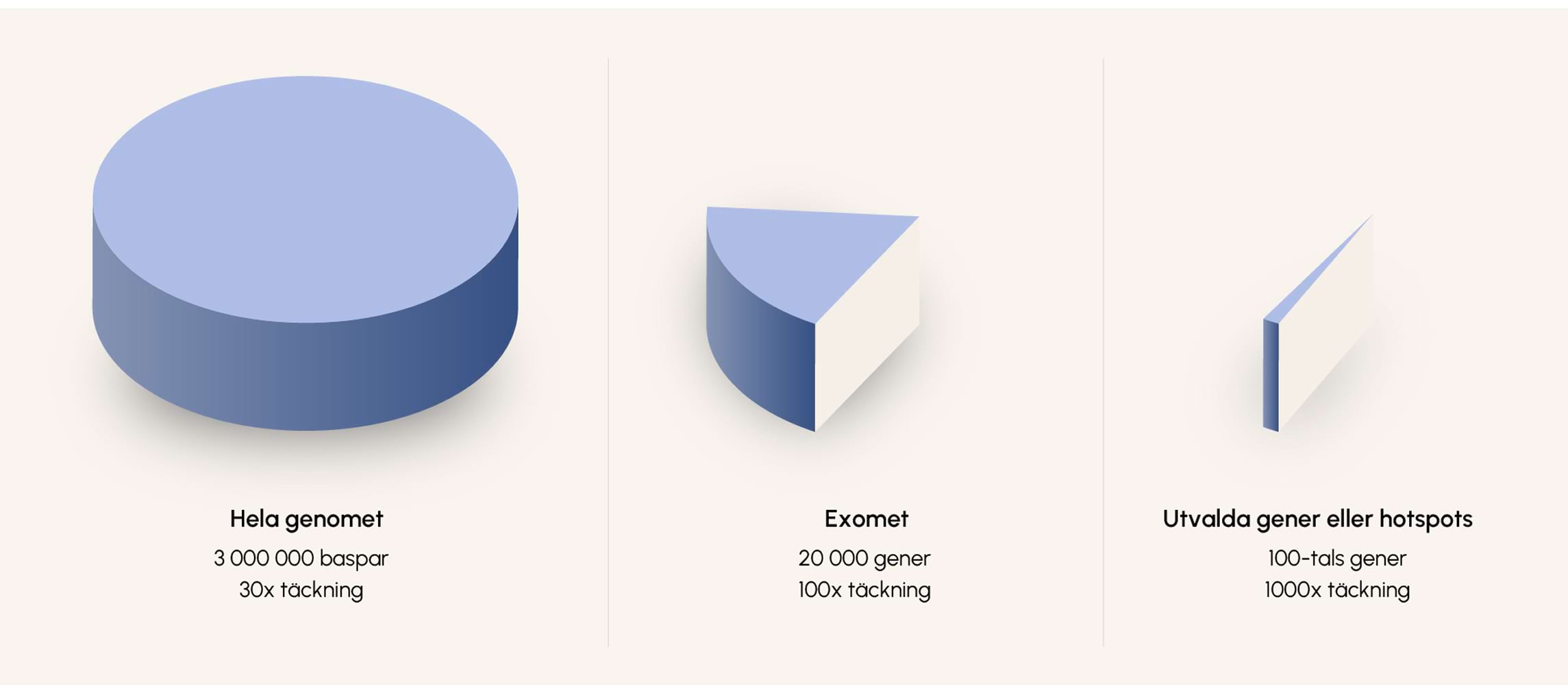
Figur 3. Det mänskliga genomet är stort och bara en liten del står för kodande sekvens (exomet). I kliniken väljs ofta ut en mindre del av exomet ut för analys.
{kind=link}
Bred analys
När hela gener eller flera gener ska analyseras används NGS (Next Generation Sequencing). Med hjälp av NGS kan man snabbt och samtidigt sekvensera stora mängder DNA eller RNA. Därför kallas teknologin ibland även MPS (Massiv Parallell Sekvensering), även om den benämningen inte fått lika stort genomslag.
- Genpaneler
En genpanel är ett urval av gener som man av olika skäl vet utgör relevanta biomarkörer. Med sekvenseringstekniker som NGS kan man analysera detta urval samtidigt i ett och samma test. Genpaneler lämpar sig när man misstänker någon av flera kända mutationer, exempelvis vid specifika cancerformer där forskningen redan pekat ut centrala drivande mutationer. Majoriteten av analyserna som utförs inom molekylärpatologin är baserade på genpaneler. Panelerna är under ständig utveckling och nya gener läggs till allteftersom man upptäcker att de har ett samband med en viss sjukdom eller av relevans för någon behandling. Man kan också använda sig av in silico-paneler (också kallade virtuella paneler eller digitala paneler) där man kan använda en bredare panel som täcker många gener men vid analysen filtrera ut endast de regioner som är av intresse för den specifika diagnosen.
- Helexomsekvensering (Whole-Exome Sequencing, WES)
Vid helexomsekvensering analyserar man samtliga proteinkodande gener, det vill säga exomet. Eftersom det är proteinkodande mutationer som oftast har direkt inverkan på proteinfunktion och därmed tumörutveckling, kan WES ge en mer omfattande bild av drivande mutationer som kan ha uppstått i en mindre känd gen än en mer riktad genpanel. Samtidigt är WES mer fokuserad och billigare än helgenomsekvensering, eftersom man inte sekvenserar den icke-kodande delen av genomet.
- Helgenomsekvensering (Whole-Genome Sequencing, WGS)
Helgenomsekvensering kartlägger hela genomet, inklusive det icke-kodande DNA:t. Detta ger en komplett bild av samtliga genetiska förändringar i tumörcellerna och kan avslöja strukturella varianter, translokationer och reglerande mutationer som annars hade missats. Nackdelarna är att metoden är mer tids- och resurskrävande samt genererar stora mängder data som kräver avancerad bioinformatik för att tolkas. För att få tillräckligt detaljerad läsning av cancergenomet krävs också väldigt mycket sekvenskapacitet för det enskilda provet vilket gör att endast ett fåtal prover kan analyseras samtidigt jämfört med riktad sekvensering. WGS är idag huvudsakligen en forskningsmetod men börjar översättas till kliniskt bruk genom translationella forskningsprojekt, till exempel det GMS-ledda initiativet inom pediatrisk cancer. GMS (Genomic Medicine Sweden) är en nationell aktör/initiativ som bland annat syftar till att integrera och harmonisera genomisk medicin i hälso- och sjukvården, samt att etablera en nationell infrastruktur för genetiska analyser.
Val av panelstorlek: Smal eller bred analys?
Vid analys av vävnadsprover för att identifiera molekylära biomarkörer behöver man ta ställning till vilken metod som ska användas. Som tidigare nämnts har olika metoder varierande styrkor, svagheter och kostnader. När det gäller riktad DNA-sekvensering uppstår frågan om man bör använda en mindre panel, fokuserad på specifika avvikelser relevanta vid exempelvis reflextestning, eller om det finns ett värde i att genomföra en bredare analys.
Fördelarna med en liten, fokuserad panel är att sekvenseringsresurserna utnyttjas optimalt; sekvenseringskapaciteten dediceras endast till de genomiska regioner som man idag vet är av intresse. En riktad analys kan göra analysen initialt mindre resurskrävande, framför allt eftersom mängden varianter som behöver tolkas begränsas.
Jämför man med det andra tillvägagångssättet, att tidigt välja en bredare panel, kan denna innehålla många gener som initialt inte är av intresse för den aktuella frågeställningen och det är dessutom tidskrävande att tolka alla varianter som hittas vid sekvensering av många gener. För att initialt fokusera analysen kan en så kallad in silico-panel användas. Detta innebär att man använder en större paneldesign, till exempel GMS560 som innehåller 544 cancerrelaterade gener, och sekvenserar samtliga dessa gener. Under den bioinformatiska analysen kan man därefter välja att endast undersöka de delar som är mest relevanta för den specifika cancerformen, vilket ofta innebär ett fåtal gener. Detta minskar resursåtgången för varianttolkning avsevärt och minskar även risken för bifynd som inte är relevanta i den aktuella cancerformen.
Reagenskostnad och arbetsinsats kan för detta tillvägagångssätt vara högre, men styrkan ligger i stället i följande faktorer:
- En större panel kan ofta möjliggöra kopietalsanalys (CNA) samt analys av andra biomarkörer som till exempel MSI och tumörmutationsbörda i samma körning, vilket ger en integrerad analys av både små (SNV/indels) och stora varianter.
- Om ytterligare information behövs i ett senare skede (till exempel inför beslut om senare behandlingslinjer), kan den bioinformatiska analysen öppnas upp för att omfatta fler gener i panelen. Det kan till exempel visa sig att tumören inte är en primärtumör och därför kräver analys av andra prediktiva markörer, eller att cancern progredierar snabbt trots första linjens behandling och en bredare analys behövs för att identifiera ytterligare behandlingsbara avvikelser. Då finns data redan tillgänglig för samtliga gener i paneldesignen.
- Nya biomarkörer identifieras kontinuerligt, och en bred paneldesign underlättar snabb implementering av nya relevanta in silico-paneler.
Sammanfattningsvis har både små och stora paneler sina respektive för- och nackdelar. Med ökande sekvenseringskapacitet och sjunkande kostnader är det dock troligt att större genpaneler kommer att bli vanligare i klinisk vardag.
Riktad analys
När man vet vad man letar efter kan man analysera enskilda gener. Vanligt använda metoder för att analysera enskilda gener är in situ-hybridisering, kvantitativ PCR (qPCR) och digital PCR.
- FISH (Fluorescerande In-Situ Hybridisering)
FISH är en metod som kan användas för att analysera större återkommande kromosomavvikelser, till exempel deletioner och amplifieringar av specifika gener eller genomiska regioner eller translokationer, alltså när en del av arvsmassan flyttats från ett ställe till ett annat. Metoden är baserad på att man låter fluorescerande prober binda in till regionerna man vill analysera och därefter undersöker kromosomerna under ett högupplöst mikroskop. På så sätt kan man upptäcka om en bit av en kromosom saknas, har duplicerats eller bytt plats.
FISH kan bland annat används vid diagnos av vissa leukemier som kännetecknas av translokationer mellan olika kromosomer, som exempelvis vid kronisk myeloisk leukemi då translokation ses mellan kromosom 9 och 22.
- qPCR (Quantitative PCR)
Kvantitativ PCR, ofta kallad real-time PCR, är en metod som inte bara amplifierar DNA eller RNA utan samtidigt mäter mängden som bildas i varje PCR-cykel. Detta sker med hjälp av fluorescerande prober som binder till den nysyntetiserade DNA-strängen och avger en signal proportionell mot mängden DNA eller RNA. Inom cancerdiagnostik kan qPCR användas för att noggrant kvantifiera genuttrycksnivåer eller detektera låga nivåer av specifika mutationer. Det kan exempelvis användas för att följa restsjukdom efter behandling genom att mäta förekomsten av en onkogen över tid.
- dPCR (Digital PCR)
Digital PCR (dPCR) är en vidareutveckling av PCR-tekniken som gör det möjligt att kvantifiera förekomsten av en viss gensekvens med extrem precision. I dPCR delas provet upp i tusentals till miljontals diskreta mikrovolymer, så att varje partition teoretiskt innehåller noll eller ett fåtal målmolekyler. Efter amplifiering avläses varje partition binärt som antingen positiv (innehåller målsekvens) eller negativ. Inom cancerdiagnostik är dPCR särskilt användbart för att mäta låga nivåer av cirkulerande tumör-DNA (ctDNA) i blod eller för att följa restsjukdom (minimal residual disease; MRD). Den höga känsligheten gör att man kan upptäcka mutationer även i en mycket liten andel av cellerna, vilket är avgörande för tidig upptäckt av återfall och för att optimera behandlingar i realtid. Detta innebär att man i teorin kan upptäcka en liten klon som bär på en resistensmekanism och justera behandlingen utifrån dennas behandlingsresponsprofil. Baksidan är att man bara kan undersöka ett fåtal enskilda mutationer i varje reaktion vilket innebär att man ofta behöver analysera provet med en bredare metod först.
DNA eller RNA
I vårt DNA finns alltså generna som utgör ritningarna för kroppens alla proteiner. Generna är uppdelade i kodande sekvens som kallas exoner och icke-kodande sekvens som kallas introner. När DNA översätts till proteiner går processen via RNA. Förenklat kan man säga att en gen på DNA-nivå skrivs av till en RNA-molekyl med både introner och exoner och därefter klipps, eller splitsas, de icke-kodande sekvenserna ut (på engelska splicing) och kvar blir ett transkript med kodande sekvens som kan skrivas av till aminosyror som sedan veckas till ett protein.
Detta leder till två egenskaper som är användbara inom molekylär diagnostik:
- Förekomsten och mängden RNA-molekyler i en vävnad motsvarar ofta proteinförekomsten även om sambandet inte är linjärt och att det förekommer scenarier då sambandet är satt ur spel. Analys av mängden RNA-molekyler kan därför användas för att skapa en genuttrycksprofil vilket till exempel används för detaljerad subgruppering av vissa bröstcancerfall.1
- Eftersom cellernas eget maskineri hjälper till att klippa bort icke-kodande sekvens kan RNA-baserade analyser utnyttjas för att hitta större strukturella varianter som ibland är omöjliga att hitta på DNA-nivå, till exempel fusionsgener som uppstått från translokationer eller alternativa transkript som uppstår av mutationer som leder till att vissa exoner hoppas över. Detta utnyttjas vid molekylär diagnostik av icke småcellig lungcancer där flera återkommande mutationer utgörs av komplexa avvikelser som säkrast hittas på RNA-nivå.
RNA analyseras idag för det mesta med hjälp av NGS-metoder som skapats för att läsa RNA, så kallad RNA-sekvensering, men beroende på situation kan utöver NGS även metoder som PCR (RT-qPCR, dPCR), NanoString och RNA-FISH också användas, antingen som komplement eller ensamt.
I arbetet med genpanelen GMS560 har det utöver DNA-analys även utvecklats en modul för analys av fusionsgener på RNA-nivå som kan utföras parallellt med DNA-analysen. Diskutera med ditt molekylärpatologiska laboratorium för att få en uppdaterad lista av fusioner som kan identifieras. På ditt laboratorium kan det även finnas andra RNA-baserade analysmetoder som fungerar lika bra.
En baksida med RNA är att molekylerna är instabila och bryts ned fort vilket gör det svårare att lagra och arbeta med dessa prover jämfört med DNA.